摘要:肠道微生物群在脓毒症发生发展中起着关键作用。然而,肠道微生物群及其代谢产物参与脓毒症过程的具体机制仍不清楚,这限制了其转化应用。本文采用微生物组学和非靶向代谢组学相结合的方法对入院的脓毒症患者的粪便标本进行分析,筛选出可能对疾病转归起重要作用的微生物群、代谢物和潜在的信号转导途径。最后,通过对脓毒症动物模型的微生物组和转录组分析对上述结果进行验证。脓毒症患者表现出共生菌群破坏和肠球菌丰度增加,拟杆菌(尤其是普通拟杆菌)负担较重的患者,APACHE II评分更高,且ICU停留时间更长。CLP大鼠肠道转录组表明,肠球菌和拟杆菌与差异表达基因具有不同的相关性,表明这些细菌在脓毒症中的作用截然不同。此外,与健康对照组相比,脓毒症患者的肠道氨基酸代谢紊乱,其中,色氨酸代谢与微生物群改变和脓毒症严重程度密切相关。肠道微生物和代谢特征的改变与脓毒症的进展相一致,有助于预测脓毒症早期患者的临床结果,并为探索新的治疗方法提供转化基础。
研究背景:脓毒症是一种严重感染导致宿主剧烈炎症反应的疾病,是一种全球范围内危及生命的疾病。在某些情况下,脓毒症会发展为多器官功能障碍综合征,导致预后恶化。越来越多的证据表明,肠道菌群失调与脓毒症等多种疾病的发病机制之间存在密切的相互作用。先前很多研究主要关注脓毒症患者的微生物特征,但在脓毒症中发生改变的细菌作用机制仍不清楚。脓毒症的代谢特征已在血清中确定,但脓毒症期间肠道的代谢变化尚不清楚。脓毒症的代谢特征已在血清中确定,但期间肠道的代谢变化尚不明确。因此,为了阐明脓毒症中肠道微生物群和代谢物的组成及其与疾病进展的关系,我们对重症监护病房(ICU)收治的脓毒症患者进行了一项观察性队列研究。入院时收集的粪便样本用于生成微生物和代谢谱。此外,我们还建立了脓毒症大鼠模型,以反映肠道基因表达特征并验证患者的微生物特征。这项研究将揭示脓毒症相关菌群的潜在生物学功能,为更早地预测和改善这种危险疾病提供理论基础。

整体研究思路
研究方法
研究设计和样本收集
本研究为描述性观察性研究,研究对象为来自中国一家中心(上海)的患者。脓毒症的定义符合第三届国际脓毒症和感染性休克共识(spesis 3)的建议。排除患有绝症的以及接受结肠造口术或回肠造口术的患者。经过测序质量过滤后,在微生物群比较中纳入了38名脓毒症患者和19名HC(图1A)。对验证组的代谢组进行单独分析,仅在该组中检测到的功能差异代谢物被确定为脓毒症相关代谢物,只有在验证组中确认的显著相关性才被定义为可靠的相关性(图1B、C)。
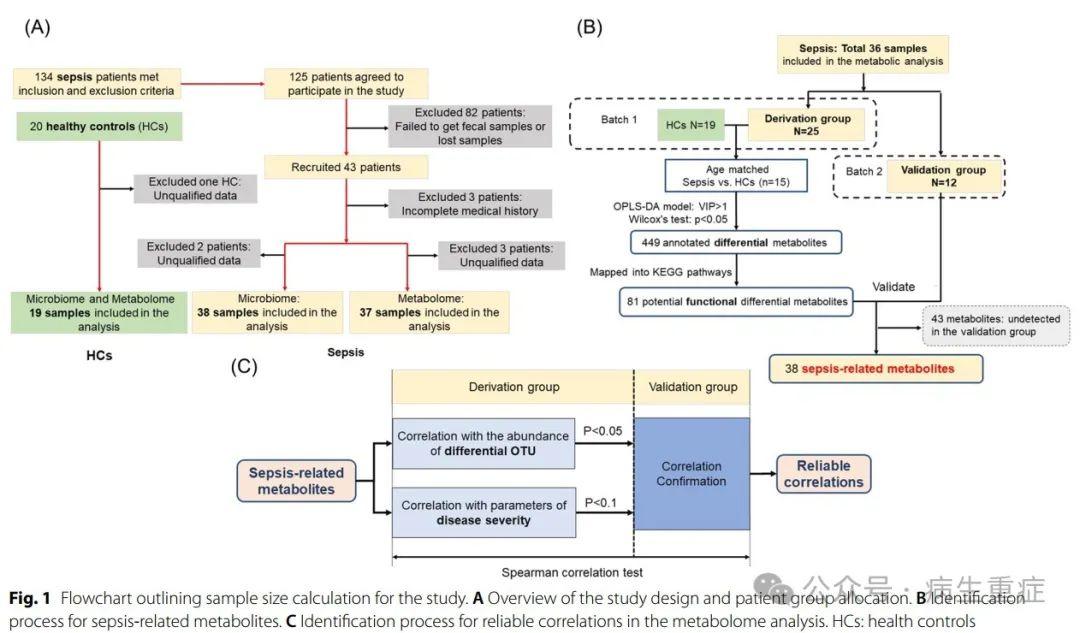
样本采集和处理
排便后立即采集患者的粪便样本,-20°C冷冻,然后转移到-80°C冰箱中长期保存。每份样本分成两部分:一部分通过16S rRNA测序进行肠道微生物组分析,另一部分通过非靶向液相色谱-质谱分析(LC/MS)进行肠道代谢组学分析。
CLP模型和RNA测序
在麻醉大鼠上建立CLP脓毒症模型,用1.5%戊巴比妥麻醉大鼠(0.5 mL/100 g体重),在腹部正中做2.5 cm的切口。在距盲肠末端1.5 cm处结扎,用14号针头穿刺两次。将盲肠放回腹腔后关闭腹壁。假手术大鼠接受相同程序,但不结扎和穿刺。在脓毒症诱导24小时后,所有大鼠均被杀死。收集含有粪便的结肠段,立即储存在−80°C,随后用于16S RNA测序以检测肠道。此外,收集从大鼠固定位置取下的远端回肠和结肠组织,用生理盐水轻轻冲洗,储存在-80°C用于转录组检测。
统计学分析
临床指标
结果连续变量以平均值和标准差表示,分类变量以频率和百分比表示。使用学生t检验比较年龄和ICU住院时间的差异。通过Fisher精确检验比较APACHE II评分≥18、SOFA评分≥10和ICU住院天数≥30的比例以及肠型之间的病死率。这些分析通过GraphPad Prism 9进行分析。
肠道微生物群
使用t检验在操作分类单元(OTU)水平比较肠道菌群的alpha多样性(Shannon和Chao指数)。进行Bray–Curtis距离度量的主坐标分析(PCoA)以反映每个样本的微生物结构,并通过相似性分析(ANOSIM)检验揭示差异。为了评估协变量对脓毒症患者菌群的潜在影响,我们通过IBM SPSS Statistics 24.0使用学生t检验或Wilcoxon检验比较各组的Shannon指数、主成分(PC)1和PC2、肠球菌OTU808载量和拟杆菌OTU773载量。从科水平到OTU水平评估线性判别分析效应大小(LEfSe),并在不同条件下设置不同的线性判别分析(LDA)评分阈值。使用Wilcoxon检验对各组间优势分类群进行比较,使用Spearman相关性分析OTU丰度与临床参数或基因表达水平之间的相关性,通过多元线性回归分析OTU773相对丰度与ICU住院天数之间的相关性。
肠道代谢物
进行Wilcoxon检验和倍数变化分析。基于VIP和Wilcoxon检验的p值(VIP的阈值>1,p<0.05)选择具有显著差异的代谢物,使用主成分分析(PCA)评估样本的交互验证。通过搜索京都基因和基因组百科全书(KEGGhttp://www。基因组。jp/kegg/)数据库来确定差异代谢物的生化途径,并根据其途径参与度进行分类。根据功能节点中代谢物的存在进行富集分析。Scipy.stats Python包用于检验由Fisher精确检验确定的富集途径的统计显著性。采用Spearman相关性分析来阐明代谢物丰度、细菌负荷和临床严重程度指标之间的关联。此外,为了评估协变量对脓毒症患者代谢组的潜在影响,我们使用IBM SPSS Statistics 24.0中的Student's t检验比较了负模式和正模式下各组的主成分(PC)1。
肠道转录组
使用Edger和DESeq2 R软件包进行差异基因表达分析,并使用带有错误发现率校正的Kal检验。经Benjamini-Hochberg校正后,用调整后的p值<0.05来鉴定差异表达基因(Deg),分析DEG的功能基团是否参与KEGG通路。生成PCA图并用于评估其相互作用的变化。为了确定核心DEG,使用相互作用基因/蛋白质检索工具(STRING)创建一个包含所有DEG的基因相互作用网络,然后将其导入Cytoscape Software 3.7.0进行可视化,选择排名靠前的DEG进一步行相关性分析。